INTRODUÇÃO:
A amiloidose é uma afecção caracterizada pelo depósito extracelular de fibrilas compostas por subunidades protéicas de baixo peso molecular. A deposição anômala da proteína amilóide promove alterações conformacionais nos peptídeos precursores, induzindo a adoção de uma estrutura beta-pregueada antiparalela, a qual apresenta elevada capacidade de auto agregação em protofilamentos fibrilares torcidos. Esse processo resulta na formação de depósitos altamente organizados, conferindo um padrão restritivo característico à cardiomiopatia amiloidótica. A manifestação clínica da doença é heterogênea, o que pode comprometer a abordagem terapêutica e o prognóstico. Destacam-se, dentre os principais subtipos, a amiloidose de cadeia leve (AL) e amiloidose associada à transtirretina (ATTR), ambas com implicações clínicas significativas.
Objetivo:
Avaliar o perfil clínico dos pacientes diagnosticados com amiloidose em dois hospitais terciários referência em cardiologia
Método:
Estudo observacional, descritivo, baseado em registro de prontuários de pacientes em acompanhamento diagnosticados com amiloidose
Resultados:
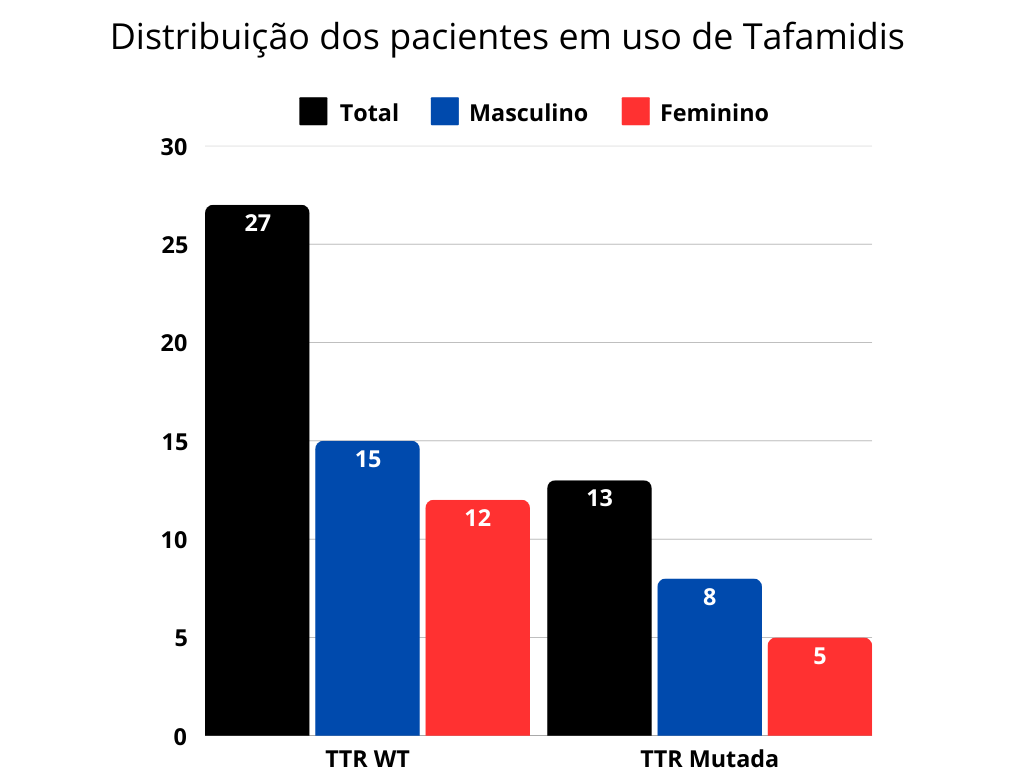
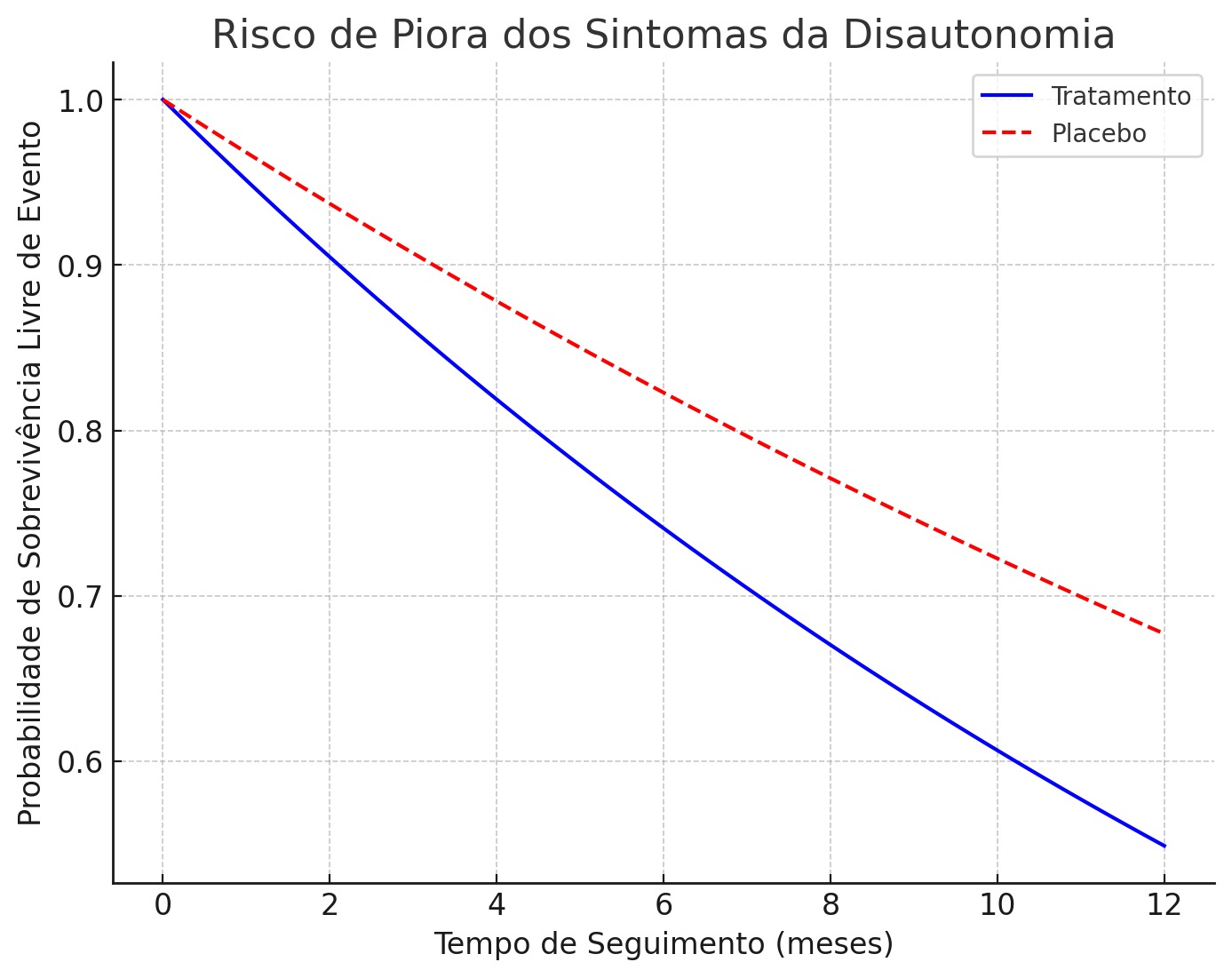
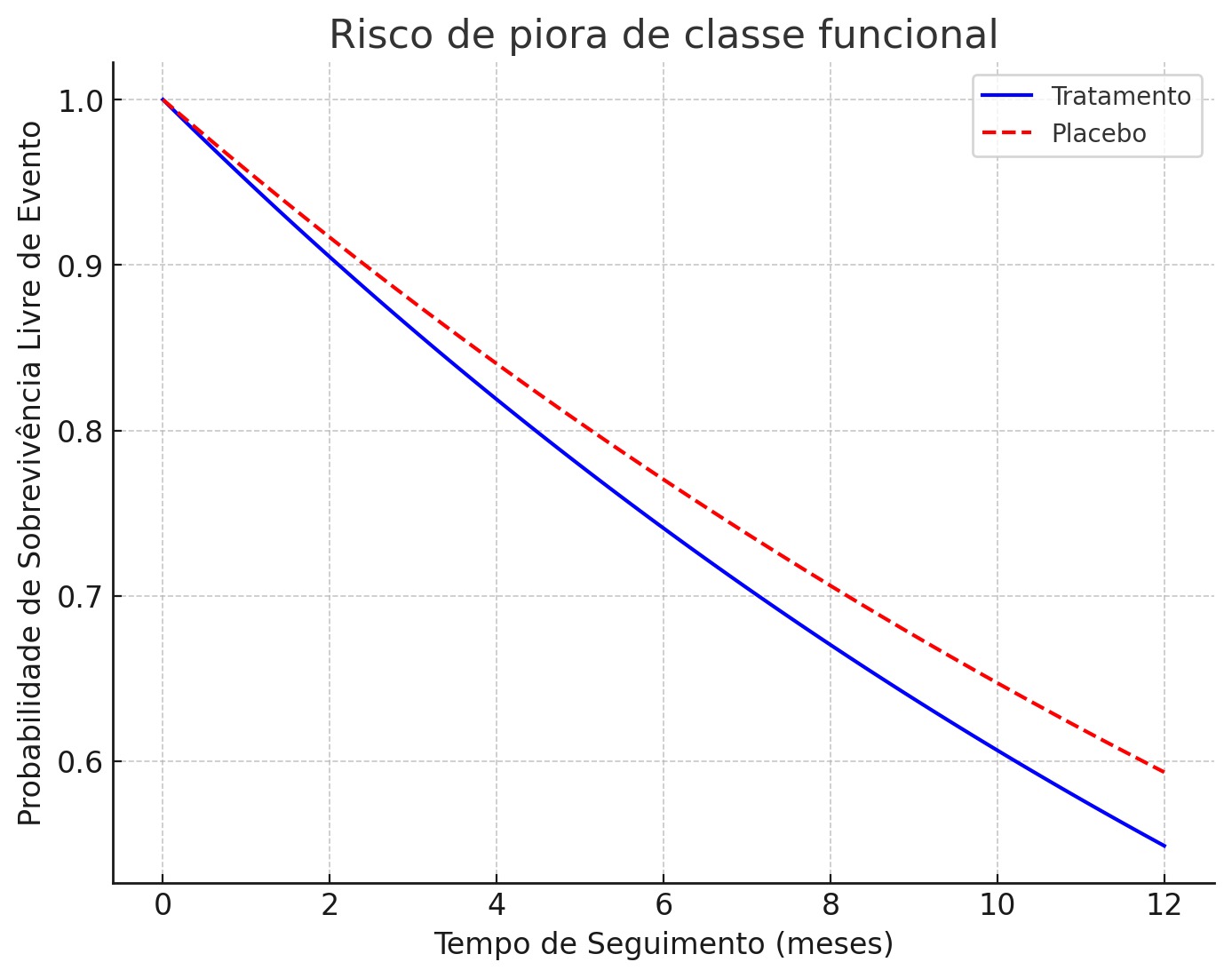
Foram incluídos 76 pacientes, dos quais 68 diagnosticados com Amiloidose TTR e 8 com Amiloidose AL. No grupo dos TTR, 38,2% tiveram mutações detectadas, sendo o marcador V122I o mais prevalente, 69,2%. A média de idade (x̅) nesse primeiro grupo fenotípico foi de 60,31 anos, com desvio padrão (σ) de 8,11 anos. 40 pacientes fizeram o uso do tafamidis, com melhora importante da classe funcional e da disautonomia. No grupo da Amiloidose AL, a média de idade (x̅) foi de 45,5 anos, com desvio padrão (σ) de 14,83 anos. 50% dos pacientes com AL foram diagnosticados com mieloma múltiplo e disfunção ventricular, sendo que 1 deles apresentou choque cardiogênico grave no momento do diagnóstico. 100% tiveram ressonância cardíaca e biópsia positivas, além de realizarem tratamento quimioterápico.
Conclusão:
A amiloidose permanece uma doença subdiagnosticada, frequentemente identificada de forma tardia, com um intervalo médio de 6 meses a 1 ano entre o início dos sintomas e a confirmação diagnóstica. Esse atraso pode comprometer significativamente o manejo terapêutico, especialmente em casos em que a deposição anômala da proteína amilóide está associada a condições subjacentes de maior gravidade, como o mieloma múltiplo. Dessa forma, torna-se essencial uma vigilância clínica rigorosa para o reconhecimento precoce dos sinais e a instituição da abordagem terapêutica mais adequada para cada paciente, visando otimizar o prognóstico e minimizar complicações.